Mucopolysaccharidosis is a rare lysosomal storage disorder with deficiency of enzymes responsible for the breakdown of glycosaminoglycans. This results in
Mucopolysaccharidosis should be suspected in patients with short stature, hepatosplenomeglay, coarse facial features, skeletal dysplasias, corneal opacities with or without neurological involvement. Diagnosis is made by doing skeletal radiographs, urinary GAG (glycosaminoglycan) levels and specific enzyme levels in the peripheral blood leukoytes.
Classification of mucopolysaccaridosis:
Mucopolysaccharidosis are classified as types I, II, III, IV, VI, VII and IX. MPS V (formerly called as Scheie syndrome) and VIII are no longer recognized.
Another classification is based on their clinical features:
- Soft tissue and skeletal involvement with or without brain disease (Type I, II, VII)
- Soft tissue and skeletal disease (MPS VI)
- Primarily skeletal involvement (MPS IVA and IVB)
- Primarily involving the brain (MPS III A-D)
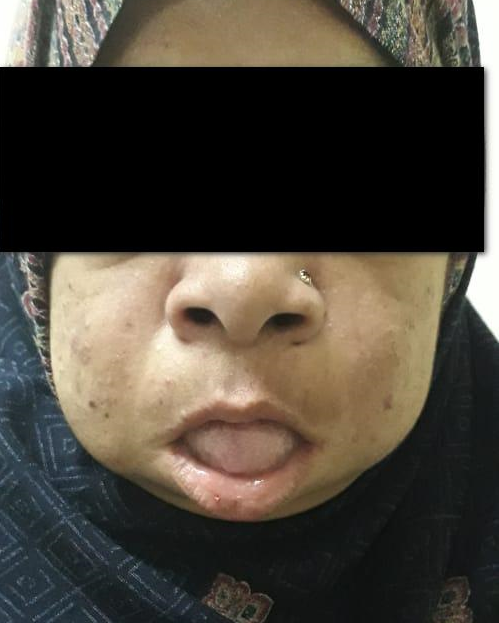
Mucopolysaccharidosis Type 1
It is an autosomal recessive disorder caused by the deficiency of lysosomal hydrolases, needed for the degradation of heparin sulfate and dermatan sulfate.
MPS 1 includes Hurler, Hurler-Scheie, and Scheie syndromes with severe, intermediate and mild clinical features of the syndrome respectively.
Hurler syndrome is the severe form and characterized by skeletal abnormalities, hepatosplenomegaly and intellectual disability. Affected infants are normal at birth, after which they develop coarse facial features, wide nasal bridge, flat midface, hepatosplenomegaly, inguinal and umbilical hernia and dysostosis multiplex.
Patients with Hurlers syndrome may have a developmental delay, recurrent respiratory tract infections and chronic recurrent nasal discharge. Patients may also develop heart failure, hydrocephalus, corneal clouding impairing vision and progressive airways narrowing.
Patients with Hurler-scheie and scheie sydromes have a less severe form of the disease. Intelligance is usually normal but skeletal deformities may be severe enough to restrict mobility.

MPS II (Hunter syndrome)
Hunter syndrome is an X-linked disorder caused by a deficiency of iduronate sulfatase. Hunter syndrome can present as mild disease or in severe form.
Hunter syndrome has similar clinical features as Hurler syndrome, however the onset of symptoms is late and patients have distinctive papular lesion over the upper arms and the scapulae.
MPS III (Sanfilippo syndrome)
It has four different forms (A, B, C and D). Patients usually present between the age f 2 to 7 years with hyperactivity, aggression, sleep disorders
MPS IV (Morquio syndrome)
These patients present at an early age with short stature, pectus carinatum, short neck and trunk, joint laxity, knocked knees,
Other manifestations include hepatosplenomegaly, corneal opacities, cervical spondylsosis, cord compression and respiratory failure.
MPS type VI (Maroteaux-Lamy syndrome)
Patients with this disorder have coarse facial features, severe skeletal disease, joint abnormalities, respiratory disease, and cardiac abnormalities (valvular disorders, such as mitral or aortic insufficiency, and less often cardiomyopathy and endocardial fibroelastosis). Untreated patients may develop obstructive sleep apnea and pulmonary hypertension.
Patients may have problems with vision and hearing and may have a developmental delay but intelligence is usually normal. Spinal stenosis and cord compression may occur.
Patients with a mild form of Maroteaux-Lamy have similar clinical features as Scheie syndrome except for short stature.
MPS type VII (Sly syndrome)
This is a very rare syndrome with severe skeletal and soft tissue abnormalities similar to MPS 1. Most of the patients are undiagnosed as they present as hydrops
MPS type IX (hyaluronidase deficiency)
This is an extremely rare disorder manifested as acetabular erosions, short stature and diffuse joint involvement.
Conclusion: Mucopolysaccharidosis is a group of rare disorders with multisystemic involvement especially the musculoskeletal system. Diagnosis is clinical and by doing urinary GAGs levels, leukocyte enzyme levels and skeletal radiographs.
The pictures given in the above article are of our patient who had corneal opacities, short stature, coarse facial features, hepatosplenomegaly, obstructive sleep apnea and cardiomyopathy.